Bugün öğrendim ki: Vücuttaki yumuşak dokuların giderek kemiğe dönüştüğü fibrodisplazi ossifikans progresifiva adı verilen nadir bir durum vardır.
Lifelı bağ dokusunun kemiğe dönüştüğü hastalık
Tıbbi durum
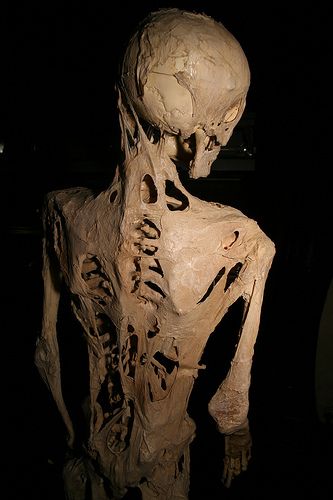
Fibrodysplasia ossificans progressivaDiğer adlarTaş adam hastalığı, Münchmeyer hastalığıFibrodysplasia ossificans progressiva hastası Harry Raymond Eastlack'ın iskeletinde karakteristik anormal kemik büyümesi görülüyor.ÖzellikTıbbi genetik, romatolojiBelirtilerSürekli kemik büyümesiGenel başlangıç10 yaşından önceSüreçYaşam boyuNedenlerDoğuştan gelen, kesin bir neden bilinmiyorFarklı tanıFibroz displaziTedaviYokİlaçPalovarotene, Sohonos ticari adıÖngörüOrta yaşam beklentisi yaklaşık 40 yıldır (düzgün yönetilirse)SıklıkDünya genelinde 801 doğrulanmış vaka (2017); milyon kişi başına 0.5 vaka oranı tahmini (2 milyondan 1'i)Ölümler60
Fibrodysplasia ossificans progressiva ( ;[1] kısaltılmış FOP), ayrıca Münchmeyer hastalığı veya daha önce myositis ossificans progressiva olarak da adlandırılan, çok nadir görülen bir bağ dokusu hastalığıdır ve kas, tendon ve bağlar gibi lifli bağ dokusunun kemik dokusuna (kemikleşme) dönüştüğü bir hastalıktır. Bilinen tek tıbbi durumdur ki bir organ sistemi başka bir organ sistemine dönüşür.[2] Tedavisi olmayan, ciddi ve sakatlayıcı bir bozukluktur.
FOP, ACVR1 genindeki bir mutasyondan kaynaklanır. Bu mutasyon, vücudun onarım mekanizmasını etkiler ve kas, tendon ve bağlar dahil lifli dokuyu, kendiliğinden veya travma sonucu hasar gördüğünde kemikleşmeye neden olur. Birçok durumda, aksi takdirde önemsiz yaralanmalar, yeni kemik oluşumu ile hasarlı kas dokusunun yerini alarak eklemlerin kalıcı olarak kaynaşmasına neden olabilir. Bu süreçte oluşan yeni kemik, "normal" kemiğe benzer, ancak yanlış yerlerde bulunur. Durumsal kanıtlar, hastalığın karakteristik kemik büyümesinden ayrı olarak eklem bozulmasına neden olabileceğini düşündürmektedir.[3]
Ekstra kemik büyümesinin cerrahi olarak çıkarılması, vücudun etkilenen bölgeyi ek kemik ile "onarmasına" neden olduğu gösterilmiştir. Kemik büyüme hızı hastadan hastanın farklılık gösterebilse de, hastalık sonunda yeni kemiğin kas dokusunu ve mevcut iskelet ile kaynaşması nedeniyle hastalar hareketsiz kalır. Bu da FOP'ye "taş adam hastalığı" lakabını kazandırmıştır.[4]
Belirtiler ve semptomlar
[düzenle]
Bilinmeyen nedenlerle, FOP ile doğan çocuklar genellikle şekil bozukluğu olan büyük ayak parmaklarına sahiptir; bazen bir eklem eksiktir veya diğer durumlarda basitçe küçük bir eklemde belirgin bir şişlik bulunur.[5] FOP kemiğinin oluşumuna yol açan ilk "şiddetlenme", genellikle 10 yaşından önce gerçekleşir.[5] Kemik büyümesi genellikle fetüslerde olduğu gibi vücudun üstünden aşağı doğru ilerler. FOP'lu bir çocuk genellikle boyundan başlayarak, omuz, kol, göğüs bölgesi ve son olarak ayaklarda ek kemikler geliştirecektir.[6]
Özellikle kemikleşme, genellikle vücudun sırt, eksenel, kraniyel ve proksimal bölgelerinde görülür. Daha sonra hastalık, ventral, apendiküler, kaudal ve distal bölgelerde ilerler.[5] Bununla birlikte, yaralanmaya bağlı şiddetlenmelerden dolayı mutlaka bu sırayla gerçekleşmeyebilir. Genellikle, hastalığın bir şiddetlenmesini karakterize eden tümör benzeri şişlikler aniden ortaya çıkar.
Şiddetlenme sırasında oluşan kemik büyümesi, etkilenmiş eklemlerde hareket kaybına neden olabilir; eğer çene/alt çene de dahil olursa, ağzı tamamen açamama, konuşmayı ve yemek yemeği sınırlandırma gibi sorunlara yol açabilir. Ayak/ayak bileği eklemlerinde bu hastalığın şiddetlenmesinin özel bir durumu, bir ayağı yere düz bir şekilde koyamama yeteneğinde sınırlamaya neden olabilir.[alıntı gerekli] Kemik büyümesi, kalça veya dizin hareketsizliğine de yol açabilir ve kişinin yürüme yeteneğini etkileyebilir. Kaburga kafesi çevresindeki ek kemik oluşumu, akciğerlerin ve diyaframın genişlemesini kısıtlayarak solunum komplikasyonlarına neden olur.[5]
Bozukluk son derece nadir görüldüğü için iki milyondan bir insanda görülebilir, hastalık kansere veya fibröze yanlış teşhis edilebilir. Bu, doktorların biyopsiler yapmasına neden olabilir ve potansiyel olarak FOP kemiğinin büyümesini hızlandırabilir.[7] FOP ile doğan kişilerde şekil bozukluğu olan ayak parmakları veya başparmakların varlığı, bu bozukluğu diğer iskelet problemlerinden ayırmaya yardımcı olur.[8]
Uygun tıbbi yönetimle ortanca yaşam yaşı 40 yıldır. Ancak, gecikmiş tanı, travma ve enfeksiyonlar yaşam beklentisini düşürebilir.[9]
Nedenler
[düzenle]
FOP, 2q23-24 kromozomunda otozomal dominant bir alelden kaynaklanır.[10] Alelin değişken bir ifadesi vardır, ancak tam penetrasyonu vardır. Çoğu vaka, üreme hücrelerinde kendiliğinden meydana gelen bir mutasyondan kaynaklanır; FOP'lu çoğu kişi çocuk sahibi olamıyor veya olmak istemiyor. Benzer ancak daha az yıkıcı bir hastalık olan fibroz displazi, zigot sonrası bir mutasyondan kaynaklanır.
Hastalığa ACVR1 geni (aynı zamanda aktifin benzeri kinaz 2 (ALK2) olarak da bilinir) mutasyonu neden olur.[10] ACVR1, BMP tip-1 reseptör olan aktifin reseptör tipi-1'i kodlar. Mutasyon, ACVR1 proteinindeki kod 206'nın argininden histidine değiştirilmesine neden olur.[11][12] Bu değişim, ACVR1'in anormal aktivasyonuna yol açarak bağ dokusunun ve kas dokusunun ikinci bir iskelete dönüşümüne yol açar. Bu, endotel hücrelerinin mezenkimal kök hücrelere, ardından kemiğe dönüşmesine neden olur.[13] Normalde, ACVR1 geni, BMP reseptörlerine (Tip I BMPR ve Tip II BMPR) kondrogenez sinyali için bağlanan aktifin reseptör tipi-1 transmembran kinazını kodlar. BMP'ler, Transforming growth factor-beta (TGF-β) proteinleri olarak bilinen bir protein süper ailesine aittir. ACVR1 proteininin BMP reseptörlerine bağlanması, gelişim sırasında endokondral kemik oluşumunu ve ayrıca iskelet ve doku homeostazını başlatan bir sinyal kaskadını başlatır.[14]
Genetik
[düzenle]
FOP, otozomal dominant bir bozukluktur. Bu nedenle, etkilenen heterozigot bir ebeveynin ve etkilenmemiş bir ebeveynin çocuğunun etkilenme olasılığı %50'dir. İki etkilenen birey etkilenmemiş çocuklar üretebilir. İki etkilenmemiş birey, genin mutasyonu sonucu etkilenen bir yavru üretebilir. Homozigot dominant form, heterozigot forma göre daha şiddetlidir.[15]
Kemiğe neden olan protein, bir fetüsün kemikleri rahimde oluştuktan sonra inhibitör bir protein tarafından normalde devre dışı bırakılır, ancak FOP'lu hastalarda protein çalışmaya devam eder. FOP'lu hastalarda anormal kemik oluşumu, travma veya büyüme yerlerinde hasarlı bağ dokusu veya kas hücrelerinin, apoptoz sırasında (kendini düzenleyen hücre ölümü) kemik onarımı için bir enzim yanlış ifadesine neden olmasıyla oluşur ve bağışıklık sistemi yanıtında sağlanan fazla kemik morfogenetik protein 4 (BMP4) içeren lenfositlere neden olur. Sonuçlanan kemik, normal iskeletten bağımsız olarak kendi ayrı iskelet elemanlarını oluşturarak oluşur. Ancak bu elemanlar normal iskelet kemiği ile kaynaşabilir.[16] Diyafram, dil ve ekstraoküler kaslar, ayrıca kalp ve düz kaslar bu işlemden muaf tutulur.[5] Yanlış enzim bağışıklık tepkisinde çözülmediği için, vücut yanlış BMP4 içeren lenfositleri sağlamaya devam eder. BMP4, normal embriyonun iskeletinin gelişimine katkıda bulunan bir üründür.[17]
ACVR1 geni, bir kemik morfogenetik protein (BMP) reseptörünü kodlar; bu gen FOP'de mutasyona uğramıştır. Kemik ve kasların büyümesi ve gelişmesinden sorumludur. Tipik R202H mutasyonu, aktivatör GS-döngüsüne daha az sıkı bir şekilde bağlanmasına neden olur.[18] Sonuç olarak, ACVR1 etkin bir şekilde kapatılmaz ve kemik ve kıkırdakta aşırı büyüme ve eklem kaynaşması meydana gelir.[19] Diğer kalıntılara ilişkin atipik mutasyonlar benzer şekilde çalışır. Bazı durumlarda, reseptör, aktivatör liganına bağlı olmaksızın aktif olduğu sinyali verebilir.[20]
FOP vakalarının çoğu yeni bir gen mutasyonunun sonucudur: bu kişilerin ailelerinde bu özel hastalığa ait bir geçmiş yoktur. Bireyin mutasyonu etkilenen bir ebeveynden miras aldığı bazı vakalar da vardır.[19]
Tanı
[düzenle]
Genellikle FOP radyografilerle teşhis edilebilir. Bu bozukluğun radyoloji yoluyla erken tanısı, biyopsiler gibi gereksiz invaziv incelemelerden kaçınmak için çok önemlidir. En küçük veya önemsiz travmalar veya kas içi enjeksiyonlar, iltihaplanma yoluyla hastalığın ilerlemesini artırabilir, bu nedenle radyolojinin tercih edilmesi gerekir. Klinikçiler, bu nadir varlığın farkında olmalıdır, çünkü genellikle kanser veya diğer iyi huylu varlıklar (örneğin enfeksiyon) ile yanlış teşhis edilir ve bu da hastalığın ilerlemesini hızlandırabilen biyopsilere yol açabilir.[21]
Patlamalar klinik olarak alkalen fosfataz ve kemik spesifik alkalen fosfataz seviyelerinde yükselme ile ölçülebilir.[22] FOP'nin başka bir ayırt edici işareti, şekil bozukluğu olan kısaltılmış büyük ayak parmağı, şekil bozukluğu olan distal birinci metatarsal ve eksik veya anormal birinci falanks ve/veya interfalangeal eklemdir.[23]
Tedavi
[düzenle]
FOP'nin tedavisi hala mevcut olmamasına rağmen, umut verici bir ilerleme onaylanan Sohonos (palovarotene) tedavisinde yatmaktadır.[24] Dikkat çekici bir şekilde, FOP'lu bir hastada kemiği cerrahi olarak çıkarma girişimleri, yeni kemiğin patlayıcı büyümesine yol açabilir.[25] Anestezi altında iken, FOP'lu kişiler intübasyon, kısıtlayıcı akciğer hastalığı ve kalbin elektriksel iletim sistemindeki değişiklikler ile karşılaşabilirler.[26] Düşme veya yumuşak doku yaralanması riskini artıran aktivitelerden kaçınılmalıdır, çünkü en küçük travma bile heterotopik kemikleşmeyi tetikleyebilir.[27]
Bozukluğun etkili kesin tedavileri olmamasına rağmen, şiddetlenmeler veya kas hasarından kaynaklanan iltihaba yanıt olarak, iltihabı bastırmak için ara sıra anti-enflamatuar ilaçlar gibi tedaviler mevcuttur. Şu anda, cerrahi işlem, insizyon veya kas veya bağ dokusuna uygulanan dikiş noktalarında hızlı kemik oluşumunu tetikleyebileceği için FOP'lu kişiler için genellikle önerilmez. Ancak, hayati önem taşıyan bir cerrahi işlem düşünülebilir; ancak cerrahi bir planın FOP uzmanı ile geliştirilmesi en iyi uygulama olabilir. Eklem kontraktürlerinin cerrahi olarak açığa çıkarılması genellikle başarısız olur ve yeni, travmaya bağlı heterotopik kemikleşme riskini taşır.[28]
Özellikle alel seçici LNA gapmerleri kullanan antisens aracılı tedavi, FOP için umut verici bir strateji haline gelmiştir[29]. Bu yaklaşım, heterotopik kemikleşmeye neden olan ve aktifin A'ya anormal yanıt veren mutasyona uğramış ACVR1 genine odaklanır. Son araştırmalarda, LNA gapmerleri patolojik ACVR1R206H transkriptinin ifadesini etkili bir şekilde azaltırken, wild-type ACVR1 genini koruduğu için FOP ile ilişkili osteojenetik farklılaşmayı seçici olarak baskıladılar. Bu yeni antisens yaklaşımı, bu ve potansiyel olarak diğer otozomal dominant bozukluklar için hedefli genetik tedavide bir atılım temsil ederek FOP'da terapötik uygulama potansiyeli sunmaktadır.
Epidemiyoloji
[düzenle]
2017 itibariyle, dünya genelinde yaklaşık 800 FOP vakası doğrulanmış ve bu da FOP'yi bilinen en nadir hastalıklardan biri yapmaktadır.[30] FOP'nin tahmini görülme sıklığı, milyon kişi başına 0.5 vaka olup tüm etnik grupları etkilemektedir.[30]
Tarihçe
[düzenle]
FOP'den etkilenen bireyleri tanımlayan tıbbi raporlar, Dr. Guy Patin'in 1692 tarihine kadar uzanmaktadır.[30] FOP, başlangıçta kas iltihabı (miyosit) nedeniyle kemik oluşumuna yol açtığı düşünülen myositis ossificans progressiva olarak adlandırılmıştır.[30] Hastalık, 1970'te kaslardan farklı yumuşak dokuların (örneğin bağlar) da hastalık sürecinden etkilendiği keşfedildikten sonra Victor A. McKusick tarafından yeniden adlandırılmıştır.[30]
En bilinen FOP vakası Harry Eastlack'indir (1933-1973). Hastalığı on yaşında başlamış ve Kasım 1973'te zatürreden 40. yaş gününden altı gün önce ölene kadar vücudu tamamen kemikleşmiş, sadece dudaklarını hareket ettirebilmişti. Eastlack, yaşamı boyunca başka bir FOP hastasıyla hiç karşılaşmadı.[31]
Eastlack, bedenini bilim için bağışladı ve iskeleti şu anda Philadelphia'daki Mütter Müzesi'nde bulunmaktadır ve FOP çalışmasında paha biçilmez bir bilgi kaynağı olmuştur. FOP'lu bir diğer kişi olan Carol Orzel (20 Nisan 1959 – Şubat 2018), aynı zamanda bedenini müzeye bağışladı ve iskeleti, Eastlack'in yanına Şubat 2019'da orada sergilendi.[32][33]
Araştırma
[düzenle]
İzotretiyonin, oral kortikosteroidlerle birlikte etidronat ve perheksilin maleatın etkinliğini gösteren klinik çalışmalar başarısız oldu, ancak hastalığın değişken seyri ve düşük yaygınlığı belirsizlik yaratmaktadır.[22]
Nadir hastalıklara odaklanan bir avuç ilaç şirketi şu anda FOP için çeşitli tedavi yaklaşımlarını araştıran farklı aşamalarda bulunmaktadır.
Ağustos 2015'te, ABD Gıda ve İlaç Dairesi (FDA) Yetişkin Ürünleri Geliştirme Ofisi, La Jolla Pharmaceuticals'a FOP için iki yeni bileşik için yetim ilaç belirlemesi verdi. Bileşikler, ACVR1 (ALK2)'yi seçici olarak engellemek üzere tasarlanmış küçük molekül protein kinaz inhibitörleridir.[39]
Ağustos 2015'te, Clementia Pharmaceuticals da FOP tedavisi için palovarotene'i inceleyen Faz II klinik çalışmasına 6 yaş ve üstü çocukların katılımını başlattı.[40] Klinik öncesi çalışmalar, palovarotene'in, BMP yolundaki ikincil haberci sistemlerini inhibe ederek hayvan modellerinde anormal kemik oluşumunu engellediğini gösterdi.[41] Clementia, palovarotene'i daha önce sağlıklı gönüllüler ve kronik obstrüktif akciğer hastalığı olan 800'den fazla kişide değerlendiren Roche Pharmaceuticals'tan lisansladı. Palovarotene, FDA tarafından Hızlı Yol belirlemesi aldı ve hem FDA hem de Avrupa İlaç Ajansı (EMA) tarafından FOP tedavisi için yetim ilaç belirlemesi aldı.[40]
Eylül 2015'te, Regeneron, aktifin A tarafından ACVR1 reseptörünün aktivasyonunu içeren hastalık mekanizması hakkında yeni bilgiler açıkladı. Şirket, 2016'da sağlıklı gönüllülerde aktifin antikoru REGN 2477'nin Faz 1 çalışmasını başlattı; 2017'de FOP hastalarında Faz 2 çalışması gerçekleştirildi.[42]
Başka bir potansiyel terapötik yaklaşım, normal ACVR1 gen ifadesini korurken mutasyona uğramış mRNA'ya yönelik alel spesifik RNA girişimini içerir.[43]
FOP'de heterotopik kemik oluşumunun mekanizmaları hakkındaki daha fazla araştırma, ekstra iskelet kemik oluşumunu içeren diğer bozuklukların tedavisinin geliştirilmesine yardımcı olabilir.
Fibro/adipojenik öncül hücreler (FAP'lar), hem FOP'ye neden olan ACVR1(R206H) mutasyonuna sahip farelerin kaslarında ve tendonlarında aktifin A bağımlı ektopik kemik oluşumundan sorumlu hastalığa neden olan hücre tipi olabilir.[44]
Aralık 2019'da, Ipsen, büyüme plaklarını erken kaynaşma raporları nedeniyle 14 yaş altındaki kişiler için kısmi klinik bir duraklama yayınladı.
Yakın zamanda, 2021 itibariyle, potent heterotopik ossifikasyon inhibitörü olarak sarakatinib, vahşi tip ve ACVR1 mutasyonlu farelerde Faz III klinik çalışmalarında bulunmaktadır.[45]
Ayrıca bakınız
[düzenle]
Uluslararası FOP Derneği - Amerikan örgütü
Osteojenez imperfekta - Kırık kemiklere neden olan genetik bozukluklar grubu, FOP'den sorumlu genlerle ilgili mutasyonlar tarafından belirlenir
İlerleyici kemik heterotopisi - Kutanöz veya subkutan kemikleşme ile karakterize nadir bir genetik durum
FOP Arkadaşları - İngiliz yardım kuruluşuWikidata açıklamalarını bir yedek olarak gösteren sayfalar
Referanslar
[düzenle]
Daha fazla okuma