Bugün öğrendim ki: "akıcı beyinli" olmanın aslında Lissensefali adı verilen bir rahatsızlık olduğu, nadir görüldüğü ve nöbetler, ciddi zihinsel/fiziksel engeller ve çok daha düşük bir yaşam beklentisi gibi birçok soruna yol açtığı, ancak tedavilerin daha iyi hale geldiği
Beyin yüzeyinde kıvrımların bulunmamasıyla karakterize bir doğum kusuru
"Agyria" buraya yönlendirilir; Argyria ile karıştırılmamalıdır.
Tıbbi durum
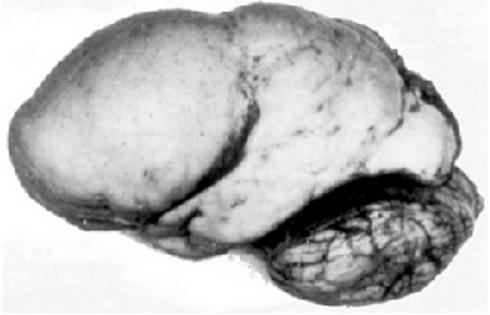
Lissensefaliİnsanda yüzey girintisi (girişim) olmayan lissensefalik beyin
UzmanlıkTıbbi genetik, nöroloji
NedenlerGirişimin olmaması
TedaviAşağıda bakınız
TahminGenellikle genç yaşta ölürler; ayrıntılar için aşağıya bakınız
Lissensefali ( , "pürüzsüz beyin" anlamına gelir)[1], beynin tümünün veya bir kısmının yüzeyinin pürüzsüz göründüğü nadir beyin bozukluklarından bir kümedir.[2] Gebeliğin 12 ila 24. haftaları arasında kusurlu nöronal göç nedeniyle oluşur, bu da beyin kıvrımlarının (giriler) ve oluklarının (sulcuslar) gelişmemesine yol açar.[3] Kafa bozukluğu türlerinden biridir. Beyin yüzeyinin görünümünü tanımlamak için agyria (giri olmadığı) ve pakiriyria (geniş giriler) gibi terimler kullanılır.
Lissensefalili çocuklar genellikle önemli gelişimsel gecikmeler yaşarlar, ancak bu gecikmeler çocuğa göre büyük ölçüde değişir, beyin malformasyonunun derecesine ve nöbet kontrolüne bağlı olarak. Yaşam beklentisi genellikle solunum problemleri nedeniyle kısalabilir.
Belirtiler ve semptomlar
[düzenle]
Etkilenen çocuklar ciddi psikomotor bozukluklar, gelişme yetersizliği, nöbetler ve kas spazmları veya hipotoni sergilerler.[4] Bozukluğun diğer semptomları arasında alışılmadık yüz görünümü, yutma güçlüğü ve ellerin, parmakların veya ayak parmaklarının anomalileri bulunur.
Lissensefali belirtileri, gebelik yaklaşık 23. haftasında ultrasonla saptanır ve prenatal MRI ile doğrulanması gerekir. Beyin yüzeyinin sulcus ve girislerinin yokluğu veya azalması ve kalınlaşmış bir korteks ile karakterizedir.[5]
İki ana lissensefali türü, Klasik (Tip 1) ve Taş Döküm (Tip 2) arasında anatomik belirtiler farklılık gösterir. Klasik lissensefali durumunda, serebral korteks kalınlaşır ve normal altı katman yerine yalnızca dört katmanı ayırt edilebilir.[5]
Taş Döküm lissensefali, kortikal yüzeyin taş dökümüne benzer veya pürüzlü görünümünden dolayı adlandırılmıştır. Bu düzensiz kortikal yüzey, tamamlanmamış organogenez nedeniyle serebral kortekste ayırt edilebilir katmanlara yol açmamasından kaynaklanmaktadır. Taş Döküm lissensefalisi, serebral korteksin gri maddesinde bir azalma ve anormallikler göstermektedir.[5]
Nedenler
[düzenle]
Lissensefali nedenleri arasında ilk trimesterde rahim veya fetüsün viral enfeksiyonları[6] veya gebelikte erken dönemde fetal beyne yetersiz kan akışı sayılabilir. Ayrıca, lissensefali için bir dizi genetik neden vardır, bunlar arasında reelin geni (7. kromozomda) [7] ve X kromozomu ve 17. kromozomdaki diğer genlerin mutasyonları bulunur. Lissensefali riski varsa genetik danışmanlık genellikle genetik testlerle birlikte sunulur.
Nöronal göç
[düzenle]
Serebral korteksin kıvrılması, genel beyin fonksiyonu ve bilişsel yeteneklerin gelişiminde önemlidir.[8] Nöronal göç, sinir sisteminin gelişimi sırasında nöronların beyindeki son konumlarına göç etme sürecidir.[9] Sinir sisteminin bu gelişimi gebelik 12 ila 16. haftaları arasında gerçekleşir.[9] Nöronlar ventriküler bölgede oluşturulur.[8] Nöronlar daha sonra radyal glia boyunca hareket ederek kortikal bölgeye ulaşırlar. Radyel ve tanjantiyel göçün bozulması, lissensefali olarak bilinen azalmış veya yok olmuş girilenlere neden olur.[10]
Serebral korteksin pürüzsüz görünümüne yol açan girilerin olmaması, sinir sisteminin gelişim aşamalarında anormal nöronal göç nedeniyledir. Lissensefali nedeni hem genetik hem de genetik olmayan faktörlerle ilişkilendirilmiştir.[11] Üç ana lissensefali tipi tanımlanmıştır ve tüm tipler benzer semptomlar sergilemesine rağmen her bir tipin patogenezi farklılık göstermektedir.[10]
Lissensefali ile ilişkili genler hala keşfedilmektedir; ancak genetik alanındaki ilerlemeler sayesinde bireysel genler lissensefali nedeni olarak tanımlanmaktadır.[12] LIS1, DCX (doublecortin), ARX (aristaless related homeobox), RELN'deki mutasyonların hepsi lissensefaliye neden olduğu belirlenmiştir.[13] Viral enfeksiyonlar da lissensefaliye neden olabilir.[14]
Bilinen genetik ve viral nedenler aşağıdadır:
LIS1
[düzenle]
LIS1 (aynı zamanda PAFAH1B1 olarak da bilinir), en çok çalışılanıdır. LIS1, 17p13.3 kromozomunda bulunur.[10] LIS1, nöronal çekirdeklerin mikrotübüller boyunca hareketinde önemli rol oynayan motor protein dinin'in düzenlenmesinde önemlidir.[11] LIS1'i içeren mutasyon veya delesyon, hem Yalıtılmış Lissensefali sendromu hem de Miller-Dieker sendromu ile ilişkilidir.[15] Ancak Miller-Dieker sendromunda, yüz ve diğer konjenital anomaliler ve kusurlara yol açan, 17. kromozomdaki bitişik genlerin ek delesyonları bulunur.[15] Bu mutasyon veya 17p13.3 kromozomunun tamamının veya bir kısmının silinmesi, LIS1'in mikrotübüller protein dinin ile etkileşen bir enzim kodlaması nedeniyle yetersiz nöronal göçe neden olur.[11] LIS1 mutasyonu veya delesyonu ebeveynlerden miras alınmaz ve bu nedenle tekrarlama olasılığı düşüktür.[10]
Bu gendeki bir mutasyona sahip ve otozomal dominant kalıtım desenine sahip bir Çin ailesi bildirilmiştir.[16]
DCX
[düzenle]
DCX veya doublecortin, gelişmekte olan nöronal süreçlerde mikrotübüllerin işlevi ve taşınması ile ilgili mikrotübüllerle ilişkili bir protein kodlayan LIS1'e benzer bir doublecortin proteinini kodlar.[12] DCX mutasyonu, serebral kortekste neokortikal katmanlaşmanın düzensizleşmesine yol açarak kıvrılmayı azaltır.[17] DCX, X kromozomunda yer alır ve bu nedenle bu mutasyon miras alınabilir, ancak yine de rastgele ortaya çıkabilir. X kromozomuyla bağlantılı bir anormallik olması nedeniyle geni miras alan erkeklerin daha ciddi şekilde etkilenme olasılığı daha yüksektir. DCX mutasyonunu miras alan kadınlarda sendromun daha hafif bir formu görülür.[11]
ARX
[düzenle]
ARX geni, birçok doku ve yapının oluşumunu kontrol etmek için erken embriyonik gelişimde aktif olan aristaless related homeobox genlerini kodlar. ARX, embriyonik ön beyinin gelişimi, nöronların göçü ve iletişimi ile internöronların göçü ve çoğalmasında yer alır.[13] ARX, gangliyonik çıkıntılar ve neokortikal ventriküler bölgede ifade edildiği için hem radyal hem de tanjantiyel göçü etkileyebilir. DCX'e benzer şekilde, ARX, X kromozomuyla bağlantılı bir gendir ve beyin bölümlerinin yokluğu, anormal genital organlar ve ciddi epilepsi gibi diğer semptomlarla ilişkilendirilmiştir.[13]
RELN
[düzenle]
Reelin (RELN), nöronal göçün düzenlenmesine yardımcı olmak üzere salgılanan ekstrasellüler bir matris glikoproteindir. Farelerde RELN eksikliğinin göç eden nöronlarda eksiklikler gösterdiği görülmüştür. Bildirilen durumlarda, RELN eksikliğinden kaynaklanan lissensefali, ön beyin bölgelerinde ve çok küçük bir serebellumda daha şiddetli olmuştur.[18]
Viral enfeksiyon
[düzenle]
Lissensefaliye, gelişmekte olan fetal beyne virüsler ve yetersiz kan akışı nedeniyle de rastlandığı kaydedilmiştir. Sitomegalovirüs (CMV), doğuştan kusurlara neden olabilen herpes ile ilişkili bir virüstür.[14] CMV, beynin gelişmekte olan germinatif matrisine yüksek bir afiniteye sahiptir. Enfeksiyonun ciddiyetinin fetüsün enfekte olduğu gebelik haftasına göre orantılı olduğu gözlemlenmiştir. Lissensefaliye yol açan erken enfeksiyondur.[14] Çünkü erken enfeksiyon, nöronların göçünü ve gelişimini bozar.[14]
Tanı
[düzenle]
Lissensefali tanısı genellikle doğumda veya doğumdan hemen sonra ultrason,[19] bilgisayarlı tomografi (BT) veya manyetik rezonans görüntüleme (MRI) ile konulur.[20] Ancak bu sonuçlar dikkatlice yorumlanmalıdır, çünkü deneyimli radyologlar bile beynin farklı bir gelişimsel malformasyonu olan polimikrogiriyadan lissensefaliyi yanlış teşhis edebilir.
Doğum öncesi, hamilelik sırasında rutin olarak yapılan karmaşık ultrasonlar, serebral bir anormalliğin varlığını gösterebilir, ancak bu tanı yönteminin genetik çalışmalar ve NMR gibi diğer yöntemlerle desteklenmesi gerekir ve aile öyküsü veya beyin malformasyonundan şüphelenilen diğer nedenler olmadıkça rutin ultrason taramalarının bir parçası olarak önerilmemektedir. Beyin yüzeyinin anormal gelişimini gözlemleyebilecek en erken gebelik haftası yaklaşık 20. haftadır, ancak 25-30. haftalarda yapılan ultrason taramaları daha yaygındır.[21] Bu zamana kadar fetal beyin normalde pürüzlü görünmektedir.[22] Lissensefali şüphesi varsa, korion villus örneklemesi bilinen bir genetik mutasyon bulunan bazı lissensefali varyantlarını test edebilir.
Sınıflandırma
[düzenle]
Lissensefali spektrumu, nörogörüntüleme ve genetik verilerin göç bozuklukları hakkında daha fazla bilgi sağlamasıyla daha iyi anlaşılabilmektedir. Spektrumu oluşturan yaklaşık 20 lissensefali tipi vardır. Henüz belirlenmemiş diğer nedenler de olası gözükmektedir.
Lissensefaliyi sınıflandırmak için farklı sistemler vardır. En önemli ayrım "klasik" (tip 1) ile "taş döküm" (tip 2) arasındaki ayrım[23] ancak bazı sistemler bu kategorilere uymayan ek formlar eklemektedir.
Aşağıda bazı lissensefali tipleri tanımlanmaktadır (OMIM numaraları mevcutsa dahil edilmiştir):
Kategori Tipler Klasik (veya Tip 1) lissensefali - 607432
LIS1: PAFAH1B1 gen mutasyonu nedeniyle lissensefali, alt bölümlere ayrılır:
tip 1 izole lissensefali (601545)
Miller-Dieker sendromu[24] (247200)
LISX1: doublecortin (DCX) gen mutasyonu nedeniyle lissensefali (300121)
diğer bilinen genetik defektler olmadan, izole, tip 1 lissensefali
Taş Döküm (veya Tip 2) lissensefali
Walker-Warburg sendromu (236670), ayrıca HARD(E) sendromu olarak da adlandırılır
Fukuyama sendromu (253800)
Kas-göz-beyin hastalığı (MEB) (253280)
Diğer tipler
LIS2: Norman-Roberts sendromu[25] (reelin gen mutasyonu, 257320)
LIS3: TUBA1A, 611603
LISX2: ARX, 300215
Mikrolissensefali (lissensefali ve mikrosefali)[26]
Tedavi
[düzenle]
Lissensefalili kişilerde tedavi semptomatiktir ve beyin malformasyonlarının ciddiyetine ve konumlarına bağlıdır. Tedavi bireyin semptomlarına göre özelleştirilir. Lissensefali için tedaviler, sendromun konjenital olduğu için semptomlarla ilgilenmektir. Konfor ve bakım ihtiyaçları için destekleyici bakım gerekebilir. Nöbetler ilaçla kontrol altına alınabilir ve hidrosefali için şant gerekebilir. Beslenme güçlüğü yaşanıyorsa gastrostomi tüpü düşünülebilir.
Lissensefali gibi nadir hastalıklar için farkındalık ve fon toplama konusunda çalışmakta olan bir dizi kuruluş vardır. Ayrıca, ilgili engellerle yaşayan bireylerin yaşam kalitesini artırmayı hedeflemektedirler. ABD'de bu kuruluşlar arasında Arc of the United States, National Organization for Rare Disorders ve March of Dimes bulunmaktadır.
Tahmin
[düzenle]
Lissensefalili çocukların prognozu, malformasyonun ve sendromun şiddetine bağlı olarak değişmektedir. Birçok birey 3-5 aylık gelişim seviyesinde kalır. Yaşam beklentisi kısadır ve birçok lissensefalili çocuk 10 yaşından önce ölecektir. Bazı lissensefalili çocuklar yuvarlanmayı, oturmayı, nesnelere ulaşmayı ve sosyal olarak gülmeyi başarabilir. Aspirasyon ve solunum sistemi hastalıkları, hastalık veya ölümün en yaygın nedenleridir.[27] Geçmişte yaşam beklentisinin yaklaşık iki yaşında olduğu söylenmekteydi. Ancak nöbet kontrolündeki ilerlemeler ve solunum sistemi hastalıkları tedavileriyle, etkilenen çoğu çocuk bu yaşın çok ötesinde yaşayabiliyor. Tedavilerdeki diğer ilerlemeler ve hizmet ve ekipmanın daha geniş kullanılabilirliği sayesinde, bazı lissensefalili çocuklar değişen derecede destekle yürümeyi ve daha önce aşırı olarak kabul edilen diğer işlevleri yerine getirmeyi başarıyor.
Ayrıca bakınız
[düzenle]
Girişim
2020 çalışmasında posterior baskın lissensefali ile ilişkili bir gen olan CEP85L
Referanslar
[düzenle]